
Colorimétrie, cinétique avec un capteur simple
Le présent article propose la réalisation d’un colorimètre simple, utilisable en Chimie pour colorimétrie ou cinétique, d’autres utilisations peuvent aussi être envisagées en Biologie (développement de levures, vitesse de sédimentation ou mesure de la turbidité d’une eau).
1) Principe, réalisation.
Le cahier des charges de départ est assez simple : matériel peu coûteux, réalisation simple, possibilité d’utiliser le dispositif de façon autonome ou par l’intermédiaire d’un système de mesures automatisé. Le montage est basé sur l’utilisation d’une LED haute luminosité rouge et d’une LDR. Le schéma de principe est le suivant :
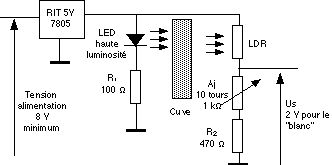
- Colorimétre schéma de principe
Quelques remarques :
![]() La LED possède un spectre assez étroit dans le rouge qu’il est possible de vérifier à l’aide d’un réseau.
La LED possède un spectre assez étroit dans le rouge qu’il est possible de vérifier à l’aide d’un réseau.
![]() La LDR est d’un type courant diamètre 10 ou 5 mm.
La LDR est d’un type courant diamètre 10 ou 5 mm.
![]() Le montage inclus un régulateur de tension 5 V, ce qui permet d’éviter tout réglage délicat avec une alimentation variable, et toute possibilité de fausse manœuvre ou de dérive pendant les mesures. La tension d’alimentation du montage doit donc être d’au moins 8 V. (Consommation de l’ensemble environ 35 mA, on peut donc envisager d’alimenter l’ensemble avec deux piles plates 4,5 V ou l’alimentation intégrée du système de mesure s’il en possède une).
Le montage inclus un régulateur de tension 5 V, ce qui permet d’éviter tout réglage délicat avec une alimentation variable, et toute possibilité de fausse manœuvre ou de dérive pendant les mesures. La tension d’alimentation du montage doit donc être d’au moins 8 V. (Consommation de l’ensemble environ 35 mA, on peut donc envisager d’alimenter l’ensemble avec deux piles plates 4,5 V ou l’alimentation intégrée du système de mesure s’il en possède une).
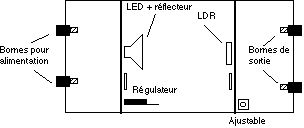
- Colorimétre - implantation
![]() L’ensemble est monté dans un boîtier type Pupicoffre 20 P de dimensions 110 x 75 x 45 en utilisant les glissières du boîtier pour positionner les deux circuits imprimés. La LED et la LDR peuvent ainsi être alignées sans problème.
L’ensemble est monté dans un boîtier type Pupicoffre 20 P de dimensions 110 x 75 x 45 en utilisant les glissières du boîtier pour positionner les deux circuits imprimés. La LED et la LDR peuvent ainsi être alignées sans problème.
![]() Il est possible de munir la LED d’un réflecteur.
Il est possible de munir la LED d’un réflecteur.
![]() La cuve est une récupération de boîte de “TicTac”. Celle-ci présente en effet un volume et des dimensions suffisantes. Les cuves à spectro ont une contenance trop faible (4 mL) ce qui pour les cinétiques chimiques pose des problèmes de manipulation. De plus, leur largeur voisine de celle de la LDR nécessite un positionnement mécanique très précis. L’emploi d’une cuve type “TicTac” permet dans certain cas d’utiliser un agitateur magnétique avec un petit aimant pour tableau d’affichage magnétique débarrassé de sa protection plastique.
La cuve est une récupération de boîte de “TicTac”. Celle-ci présente en effet un volume et des dimensions suffisantes. Les cuves à spectro ont une contenance trop faible (4 mL) ce qui pour les cinétiques chimiques pose des problèmes de manipulation. De plus, leur largeur voisine de celle de la LDR nécessite un positionnement mécanique très précis. L’emploi d’une cuve type “TicTac” permet dans certain cas d’utiliser un agitateur magnétique avec un petit aimant pour tableau d’affichage magnétique débarrassé de sa protection plastique.
![]() Le couvercle du boîtier est découpé aux dimensions de la cuve. Il est également percé d’un trou permettant le passage d’un tournevis pour régler l’ajustable. Celui-ci est soudé côté piste pour permettre son réglage sans introduire de lumière parasite. En fonctionnement l’ensemble est recouvert d’un autre petit boîtier permettant d’éliminer les lumières parasites.
Le couvercle du boîtier est découpé aux dimensions de la cuve. Il est également percé d’un trou permettant le passage d’un tournevis pour régler l’ajustable. Celui-ci est soudé côté piste pour permettre son réglage sans introduire de lumière parasite. En fonctionnement l’ensemble est recouvert d’un autre petit boîtier permettant d’éliminer les lumières parasites.
![]() Avant toute manipulation, il est nécessaire de laisser le colorimètre atteindre son équilibre de fonctionnement (dérive en température de la LED, de la LDR et du régulateur intégré). Pour cela, mettre le boîtier sous tension (8 V minimum), remplir la cuve d’eau (attention à la formation éventuelle de petites bulles), couvrir l’ensemble du boîtier cache, attendre 5 minutes environ. En glissant le cache de façon à ne découvrir que l’orifice de réglage, régler l’ajustable de façon à obtenir une tension de 2 V en sortie.
Avant toute manipulation, il est nécessaire de laisser le colorimètre atteindre son équilibre de fonctionnement (dérive en température de la LED, de la LDR et du régulateur intégré). Pour cela, mettre le boîtier sous tension (8 V minimum), remplir la cuve d’eau (attention à la formation éventuelle de petites bulles), couvrir l’ensemble du boîtier cache, attendre 5 minutes environ. En glissant le cache de façon à ne découvrir que l’orifice de réglage, régler l’ajustable de façon à obtenir une tension de 2 V en sortie.
2) Colorimétrie avec des solutions de permanganate de potassium.
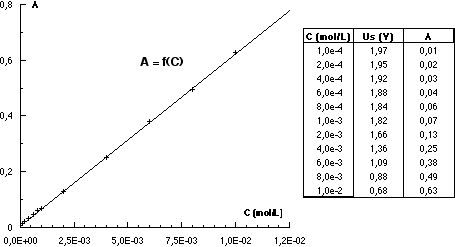
Il suffit de préparer des solutions de permanganate de potassium de concentrations données, ou de diluer progressivement une solution donnée (voir annexe 2). Dans chaque cas on relève la tension Us pour une concentration donnée. On obtient alors le tableau de mesures et le graphique suivants :

- Courbe d’étalonnage
- Détermination de concentration de solution de permanganate de potassium
Avec des concentrations comprises entre 10-2 et 10-4 mol.L-1, la fonction de transfert de ce colorimètre n’est pas linéaire, mais peut être assimilée à une fonction du second degré avec une très bonne corrélation. Cependant, si on travaille avec des concentrations inférieures à 5.10-3 mol.L-1 on peut estimer que cette fonction de transfert est linéaire dans ce domaine.
Quel que soit le domaine, il est possible de déterminer à partir de cette courbe d’étalonnage, la concentration d’une solution inconnue, en la diluant au besoin si celle-ci est trop concentrée.
Ce colorimètre a été réalisé en plusieurs exemplaires, les différences observées entre les caractéristiques des différents modèles sont minimes.
3) Vérification de la loi de Beer Lambert.
À partir des mesures d’étalonnage réalisées précédemment, il est possible de vérifier la loi de Beer Lambert.
Une LDR est telle que sa résistance est très importante (1 MΩ) lorsque l’éclairement est nul, elle est au contraire de l’ordre de quelques Ohms lorsque celui-ci est important. On peut donc poser que la résistance de la LDR est liée au flux reçu par celle-ci - pour une longueur d’onde donnée - par une relation du type R = K/F, K étant une constante caractéristique de la LDR. (Il faudra bien sûr vérifier cette hypothèse - voir annexe 3).
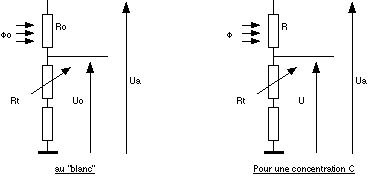
Au blanc, la LDR reçoit un flux Fo, sa résistance est alors Ro et la tension de sortie du colorimètre est alors telle que :
Rt/Uo = (Ro + Rt)/Ua d’après la relation des ponts diviseurs (Rt étant la résistance talon fixée par R2 et l’ajustable, Ua la tension d’alimentation de l’ensemble).
Soit : Ro = Rt.(Ua/Uo - 1)
Pour une concentration C de la solution de permanganate de potassium, la relation devient alors : R = Rt.(Ua/U - 1)
L’absorbance de la solution de concentration C est telle que A = log (Fo / F) = log (R / Ro) puisque R = K / F.
On obtient alors : A = log [(Ua/U - 1)/(Ua/Uo - 1)].
Comme Ua = 5 V et Uo = 2 V, la relation devient :
A = log [(5/U - 1)/(5/2 - 1)] soit finalement A = log (10/3.U - 2/3).
Il suffit de compléter le tableau d’étalonnage et de tracer la courbe A = f(C).
4) Cinétique de décoloration ions permanganate / ions oxalate.
Par interfaçage, ou manuellement, il est possible de suivre la réaction ion permanganate / ion oxalate, réaction présentant un phénomène d’auto-catalyse par les ions Mn2+.
L’équation bilan de la réaction est :
2 MnO4- + 5 C2O42- + 16 H+ ––––> 2 Mn2+ + 8 H2O + 10 CO2
Si on veut que la décoloration soit totale il faut opérer avec un excès d’ions oxalate. Pour éviter la formation de bulles de dioxyde de carbone qui viennent perturber les mesures, les concentrations doivent être faibles et enfin, le milieu doit être suffisamment acidifié (pour éviter la formation de MnO2).
Les cuves “TicTac” ayant une contenance de 20 mL, si on réalise des mélanges équivolumiques de 10 mL de chacune des solutions, il est possible de travailler avec des solutions de permanganate de potassium à partir de 2.10-2 mol.L-1. Pour opérer avec un excès d’ions oxalate, il faut alors que les concentrations soient liées par la condition Vo.Co > 5/2.Vp.Cp (p = Permanganate, o = Oxalate), soit avec des mélanges équivolumiques Co > 5/2.Cp.
Dans un premier temps, il suffit de relever les points expérimentaux U = f(t). On dispose (paragraphe 2) de la fonction de transfert [MnO4-] = f(U). Après traitement des données on obtient alors le type de courbe [MnO4-] = f(t) caractérisant bien le phénomène d’auto-catalyse.

5) Formation de soufre colloïdal. ion thiosulfate / solution chlorhydrique.
Équation-bilan de la réaction :
S2O32- + H3O+ ––––> HSO3- + S + H2O
L’étalonnage :
Celui-ci se fait en supposant que tout le soufre est formé, on prélève alors un certain volume de solution que l’on remplace par de l’eau pour avoir des solutions renfermant successivement 100% puis 90 %... de soufre par rapport à la quantité maximale. Mais cet étalonnage se fait alors que les micelles de soufre ont une certaine taille, (la réaction est arrivée à son terme), or cette taille est inférieure au début de la réaction, l’absorption n’est donc pas la même.
L’étalonnage donne les résultats suivants, la fonction de transfert étant là aussi du second degré.
Quelques précautions :
![]() Opérer avec un excès d’ions H3O+ par rapport aux ions thiosulfate
Opérer avec un excès d’ions H3O+ par rapport aux ions thiosulfate
![]() Agiter le milieu pour éviter le dépôt du soufre (le colorimètre peut être posé sur un agitateur magnétique en utilisant un petit aimant pour tableau).
Agiter le milieu pour éviter le dépôt du soufre (le colorimètre peut être posé sur un agitateur magnétique en utilisant un petit aimant pour tableau).
![]() La concentration de la solution de thiosulfate de sodium ne doit pas être trop élevée pour éviter un dépôt de soufre malgré l’agitation. En pratique Ct <= 5 10-2 mol.L-1 et donc si on a un excès d’ions H3O+, il faut donc Ca >= Ct si on opère avec un mélange équivolumique (a = acide, t = thiosulfate).
La concentration de la solution de thiosulfate de sodium ne doit pas être trop élevée pour éviter un dépôt de soufre malgré l’agitation. En pratique Ct <= 5 10-2 mol.L-1 et donc si on a un excès d’ions H3O+, il faut donc Ca >= Ct si on opère avec un mélange équivolumique (a = acide, t = thiosulfate).

Après traitement des données, on obtient la courbe suivante avec une solution 5.10-2 mol.L-1 de thiosulfate de sodium et une solution 10-1 mol.L-1 d’acide chlorhydrique.
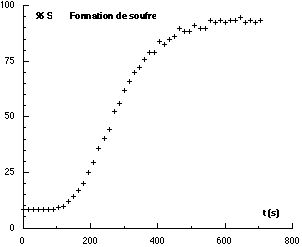
La quantité de soufre formée au début de la réaction est insuffisante pour être détectée. Néanmoins il est possible de faire des comparaisons entre des réactions faisant intervenir des concentrations différentes de solution chlorhydrique. (Il est souhaitable de garder la même concentration pour la solution de thiosulfate de sodium afin de ne pas refaire l’étalonnage).
6) Formation de diiode. peroxodisulfate de sodium / iodure de potassium.
L’équation bilan de la réaction est :
S2O82- + 2 I- ------> I2 + 2 SO42-
Il faut opérer de façon à ce que le mélange réactionnel présente toujours un excès d’ions iodure afin d’éviter la précipitation du diiode I2, précipitation qui vient perturber les mesures même en agitant le mélange réactionnel.
Les proportions stœchiométriques sont telles que Vi.Ci = 2.Vp.Cp (i = iodure, p = peroxodisulfate). Si on veut empêcher la précipitation et que l’on opère à mélange équivolumique, la condition devient alors Ci >= 4.Cp de façon à être largement excédentaire en ions iodure.
L’étalonnage se fera avec le même principe que pour le soufre. Une fois la réaction achevée, on prélève un volume donné de solution que l’on remplace par de l’eau. Il est préférable d’opérer avec la même concentration en ions peroxodisulfate pour ne pas avoir à refaire cette opération.
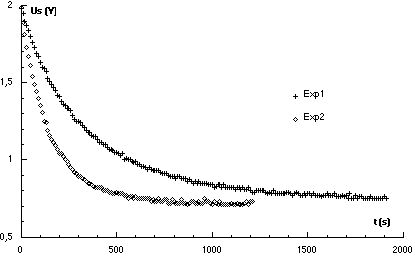
Voici par exemple deux courbes Us = f(t) obtenues dans les conditions suivantes :
peroxodisulfate de sodium // iodure de potassium
Exp1 0,1 mol.L-1 // 0,5 mol.L-1
Exp2 0,1 mol.L-1 // 1 mol.L-1
(Les données n’ont pas été traitées pour obtenir la courbe [I2] = f(t)).
Notes
Les mesures par interfaçage ont été réalisées avec Orphy GTS (voie de calibre 2 V).
Bibliographie
Étude photométrique d’une réaction lente R. Gourdiole BUP 664 p 1051
Annexe 1
À titre indicatif voici les typons des deux parties du colorimètre ainsi que les schémas d’implantation correspondants.
Pistes côté cuivre :

Implantation des composants :

Une amélioration du montage pourrait être faite en offrant la possibilité de changer la LED émettrice. On pourrait alors disposer de lumières de différentes couleurs. Ceci pose quelques petits problèmes mécaniques. On peut si on dispose de boitiers assez long, placer la LED seule sur une plaque époxy indépendemment du régulateur. Cette plaque venant s’alimenter à l’aide de cosses de type “faston” sur la plaque alimentation. Il faut en effet garantir l’alignement de l’ensemble LED, LDR.
Annexe 2
Pour l’étalonnage des solutions on peut opérer par prélèvement d’un certain volume de solution et remplacement par le même volume d’eau.
Si on dispose à un moment donné de la procédure d’un volume V de solution de concentration Ci, on prélève un volume Vp de cette solution que l’on remplace par de l’eau pour obtenir le même volume de solution de concentration Cf. La conservation de la matière permet d’écrire la relation Ci.(V - Vp) = Cf.V soit Vp = V.(1 - Cf/Ci).
Si initialement la solution avait une concentration C, à un moment donné on passe d’une solution de concentration Ci = k.C, à une solution de concentration Cf = k’.C. Si on désire que k - k’ = 0.1, la relation devient alors Vp = 0,1.V/k.
Pour un volume initial de 20 mL (ce qui est le cas de nos expériences avec les cuves type “Tic-Tac”) on obtient alors le tableau suivant, indiquant les volumes successifs à prélever et à remplacer par de l’eau.
| C | k | Vp (mL) |
| 0,9.C | 1 | 2,0 |
| 0,8.C | 0,9 | 2,2 |
| 0,7.C | 0,8 | 2,5 |
| 0,6.C | 0,7 | 2,9 |
| 0,5.C | 0,6 | 3,3 |
| 0,4.C | 0,5 | 4,0 |
| 0,3.C | 0,4 | 5,0 |
| 0,2.C | 0,3 | 6,7 |
| 0,1.C | 0,2 | 10,0 |
Annexe 3
Pour vérifier la loi de Beer Lambert, nous avons admis que la résistance de la LDR était liée au flux lumineux par une relation du type R = K/F. Le fait d’avoir bien vérifié la loi de Beer Lambert suffirait pour admettre la validité de cette hypothèse quand au comportement de la LDR. Mais on peut essayer de la vérifier directement.
Si on a bien R = K/F alors R = K.s/Pr si s est l’aire de la LDR et Pr la puissance reçue par celle-ci.
La puissance reçue par la LDR située à une distance d de la LED qui émet une puissance Pe est alors :
Pr = Pe.s/(4.pi.d2)
La relation R = K.s/Pr devient alors R = K.s.4.pi.d2/(Pe.s) = K’.d2
La résistance de la LDR doit donc dépendre de la distance d entre la LED et la LDR suivant une loi d’ordre 2.
Pour vérifier cette loi, il suffit de monter une LED et une LDR sur un banc et de mesurer la résistance de la LDR pour des distances différentes. (On peut bien sûr veiller pour cela à utiliser la même tension d’alimentation pour la LED que celle utilisée dans le colorimètre).
Les résultats obtenus sont les suivants.
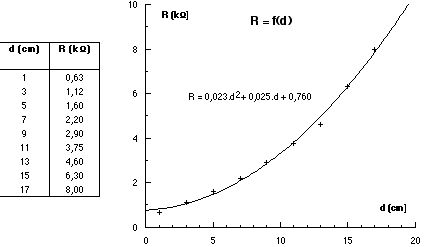
L’hypothèse est donc bien vérifiée.
Remarque : La distance d doit être mesurée à partir d’une position arbitraire, la “vraie” distance correspond au minimum de la courbe. D’autre part la résistance n’est pas nulle lorsque la distance est minimale. La relation n’est pas tout à fait du type R = K/F mais R = Rm + K/F. Ceci introduit un petit terme correctif mais qui peut être négligé pour ne pas trop alourdir les calculs avec les élèves.
Il me semble que cette vérification doit être faite, or cette précaution n’est pas prise dans certains articles proposant la vérification de la loi de Beer Lambert. La fonction logarithme dans la définition de l’absorbance “écrasant” les données, la linéarité peut sembler vérifiée. Avec les résultats obtenus à l’aide de ce colorimètre, la courbe A = log(U/Uo) = log(U/2) par exemple donne aussi une droite (sauf pour la concentration la plus élevée).